HARP-X: Healing of Antibiotic Refractory Pouchitis with Xeljanz
HARP-X is a prospective multi-centre, open label induction and randomized maintenance pilot study of Tofacitinib in subjects with chronic, antibiotic refractory or dependent pouchitis. The primary objective is to determine the effect of tofacitinib maintenance therapy on endoscopic response in subjects with active, chronic, antibiotic dependent or refractory pouchitis.
Background
Ulcerative colitis (UC) is a chronic inflammatory condition that causes continuous mucosal inflammation of the colon. Observational studies have shown increased incidence and prevalence of UC since the mid-20th century, especially in the western world. Despite medical therapy, approximately 30% of patients with UC will eventually require a colectomy, or removal of the entire colon. Restorative proctocolectomy with ileal pouch-anal anastomosis (IPAA) has become a part of standard surgical treatment for patients with refractory UC who undergo colectomy. Pouchitis, a nonspecific inflammatory disease of the ileal pouch reservoir, is the most common long-term complication in patients with IPAA, and significantly affects patient quality of life. While a majority of patients with pouchitis respond to treatment with antibiotic therapy, approximately 10%-15% of will develop chronic antibiotic-dependent or refractory pouchitis. There are currently no licensed treatments for refractory pouchitis and research into alternative therapies is needed.
Pouchitis is an inflammatory condition of the ileal pouch reservoir affecting up to 70% of patients following colectomy for UC. Symptoms of pouchitis may include diarrhoea with or without blood, urgency, abdominal pain, fevers, as well extraintestinal manifestations such as joint pain. Currently the aetiology and pathogenesis of pouchitis are not entirely clear. One line of thinking suggests that pouchitis is a recurrence of the underlying UC disease within the neorectum. Although the pouch is constructed only from the ileum, over time, villi are lost or blunted and crypts develop to form a more colon-like morphology, also known as colonic metaplasia. Associated with these changes is the presence of a chronic inflammatory cell infiltrate (e.g. lymphocytes, plasma cells, eosinophils, and histiocytes) within the lamina propria. Pouchitis is defined by acute inflammatory changes superimposed on the chronic changes with or without the presence of ulceration and crypt abscesses, and this is analogous to an acute UC flare. Furthermore, evidence suggests that an abnormal mucosal immune response to altered microflora in the pouch leads to acute and/or chronic inflammation. As in UC, there are abnormalities in immunoregulatory cytokines such as interleukins, as well as an excess of pro-inflammatory cytokines such as TNF-α. The role of an abnormal regulation of the mucosal immune system is underscored by the fact that pouchitis rarely occurs in patients with familial adenomatous polyposis (FAP), who underwent an identical surgery of colectomy with IPAA.
As such, various data point towards the option of an immunosuppressive approach using biologics such as anti-TNF agents in those patients with pouchitis that do not respond to treatment with antibiotic therapy. Patients are considered to be antibiotic dependent when the disease requires long-term, continuous antibiotic therapy to maintain remission. Those with antibiotic-refractory pouchitis fail to respond to antibiotics altogether. Both of these groups may require alternative treatment options such as oral or topical 5-aminosalicylates, corticosteroid therapy, immunomodulator therapy, or even biologic therapy. Despite there being some evidence showing efficacy in treating antibiotic refractory or dependent pouchitis with the above, many patients do not have a response to these therapies or have a response that is not sustained. The existing studies have been limited in terms of size, and additional trials looking at new treatment options are needed to increase efficacy rates in managing pouchitis.
Study Design
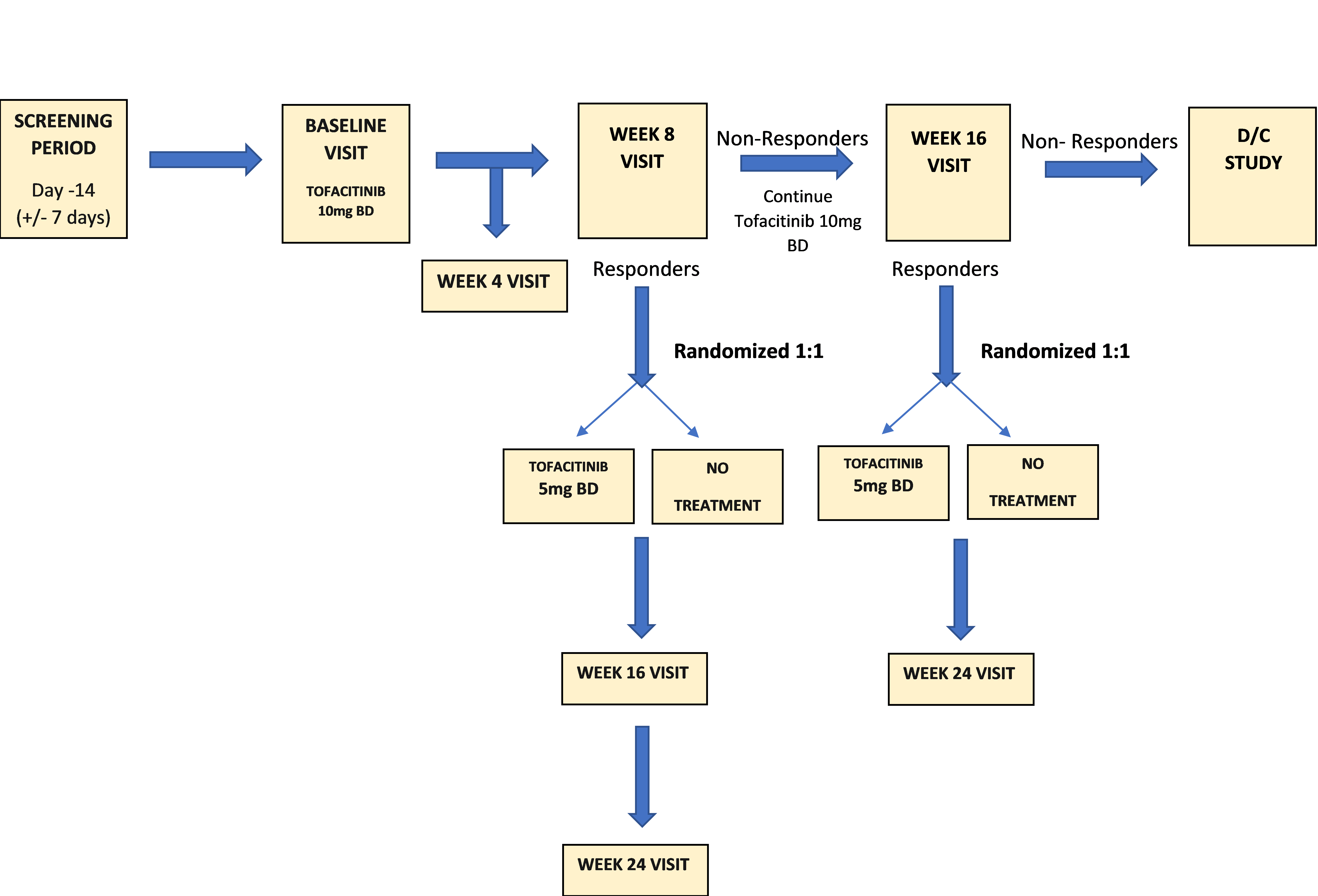
Subjects will undertake a 2-week (+/- 7 day) screening period to provide baseline data and be assessed for eligibility. At baseline visit (day 1) subjects will be treated with open label tofacitinib at the approved induction dose of 10mg BD po for 8 weeks.
Subjects will be assessed at week 8 via clinical symptoms and pouchoscopy. Responders will be randomized 1:1 to discontinue therapy (no maintenance)*, or to continue tofacitinib at the approved maintenance dose of 5mg BD po. Those with inadequate response at week 8 will continue open label treatment (10mg BD po) for an additional 8 weeks. Week 8 inadequate responders who receive an additional 8 weeks of treatment will be assessed at week 16 via clinical symptoms (pouchoscopy at this time point will be left up to the discretion of the investigator). Week 16 responders will be randomized into the maintenance arm 1:1 to discontinue therapy (no maintenance)*, or to continue tofacitinib at the approved maintenance dose of 5mg BD po. Week 16 non-responders will be discontinued from the study.
*Subjects who experience disease worsening after randomization will have a pouchoscopy for primary endpoint analysis and will be discontinued from the study. They may be re-induced with tofacitinib outside the confines of the study if the investigator deems this appropriate.
Following the baseline (day 1) visit, subjects will return to the clinic for safety and efficacy assessments at week 4, 8, 16, and 24.
Primary Aims:
To determine the effect of tofacitinib maintenance therapy on endoscopic response in subjects with active, chronic, antibiotic dependent or refractory pouchitis
Secondary Aims:
• To determine the ability of tofacitinib to improve the clinical symptoms associated with antibiotic dependent or refractory pouchitis
• To determine the effect of tofacitinib on health-related quality of life
• To determine the effect of tofacitinib on reducing pouchitis “rescue” intervention
• To determine the effect of tofacitinib on changes in biological markers including C-reactive protein, fecal calprotectin and histology
• To evaluate the duration of effect following cessation of therapy
Feasibility Aims:
• To determine the proportion of eligible patients willing to be enrolled
• To determine the proportion of eligible patients willing to be randomised
• To determine dropout rates
• To evaluate protocol specific adherence rates
• To evaluate treatment specific compliance rates
Inclusion Criteria:
1. Subjects who are willing and able to provide written informed consent
2. Male or female subjects, >/=18 years of age who
have undergone an ileo-anal pouch anastomosis (IPAA) for UC
3. Documented evidence of active pouchitis – must have a pouchitis activity score (PAS) >/= 13
4. Pouchoscopy at screening, or within the previous 3 months, documenting endoscopic activity by photography and confirmed by histology (Mayo endoscopic sub score of >/1)
5. Symptomatic activity: subjects must demonstrate increased stool frequency compared to what is considered
“normal” after their IPAA operation (“baseline”); stool frequency must be an absolute value of > 6 stools per day, and > 3 stools per day above the post-IPAA “baseline”
6. Must have chronic antibiotic dependent or refractory pouchitis
Exclusion Criteria:
1. Subjects who are unwilling or unable to give informed consent
2. Lack of effective contraception
3. Women who are pregnant or breastfeeding
4. History of allergy to tofacitinib or any of its components
5. History of regular NSAID use
6. Oral 5-aminosalicylate (5-ASA) compounds; exclude subjects who have discontinued or changed doses of oral 5-ASA within 2 weeks of the screening visit (ie: if subject is taking oral 5-ASAs dose must be stable for 2 weeks prior to screening)
7. Oral budesonide >6mg/day is not permitted (doses </= 6mg must be stable for 2 weeks prior to screening visit)
8. Oral steroids other than budesonide; exclude subjects who exceed a daily dose of 20mg prednisolone or equivalent (doses </= 20mg must be stable for 2 weeks prior to the screening visit)
9. Use of rectal compounds is not permitted; these agents must be discontinued at the screening visit
10. Immunosuppressant therapy (azathioprine, 6-mercaptopurine, methotrexate, cyclosporine, tacrolimus) is not permitted within 4 weeks of the baseline visit
11. Biologic agents (anti-TNF therapy, anti-CD20 monoclonal antibodies and anti-integrins (ie: Vedolizumab)) are not permitted within 8 weeks of the baseline visit. Ustekinumab (and all interleukin (IL)-1R antagonists, IL-6R antagonists, IL-17 antagonists, IL-12/IL-23 antagonists) are not permitted within 12 weeks of the baseline visit. Sites are allowed to assay drug levels to shorten the washout periods. The washout period can be reduced if serum levels of the biologic in question are undetectable.
12. Antibiotic therapy is not permitted during screening or any time during the duration of the study; antibiotics must be stopped prior to the screening visit
13. All other agents targeted to pouchitis, including experimental agents, must be discontinued at least 8 weeks prior to the screening visit, or for a period equivalent to 5 half-lives of the agent (whichever is longer)
14. Anastomotic stricture
15. Those unable to undertake endoscopic evaluation
16. Fecal incontinence due to anal sphincter dysfunction
17. Infections with cytomegalovirus or Clostridium Difficile
18. Intestinal malabsorption
19. Pancreatic insufficiency
20. Subjects with severe moderate to severe hepatic impairment
21. Subjects with severe renal impairment (defined as a creatinine clearance < 30mL/min)
22. Suspected irritable pouch syndrome
23. Subjects with only cuffitis (inflammation of the rectal cuff of the pouch); subjects with active antibiotic dependent or refractory pouchitis as the predominant condition, but who also have cuffitis, may be enrolled
24. Crohn’s disease of the pouch
25. Subjects with a history of neoplastic disease, except for basal cell carcinoma or non-metastatic squamous cell carcinoma of the skin
26. Subjects with latent or untreated tuberculosis, chronic viral hepatitis, recurrent herpes zoster infection, or other chronic infection likely to be exacerbated by tofacitinib therapy
27. Subjects with a history of clinically significant and/or persistent illness, which in the investigator’s opinion, would exclude entry into the trial
28. Subjects with any laboratory tests considered clinically significant at screening
29. Subjects who may be unavailable for the duration of the trial, are likely to be non-compliant with the protocol, or who are felt to be unsuitable by the investigator for any other reason
30. Subjects who have insufficient protection against the varicella zoster virus (VZV) as 1) evidenced by undetectable serum VZV antibodies and 2) who have not received varicella vaccination; subjects who have detectable varicella immunity on blood testing or who have received varicella vaccination will be considered eligible
31. Subjects in recent receipt of live vaccinations within 4 weeks prior to enrolment
The treatment period will include a 2-week screening period, a 8 week induction period, and a 16 week follow-up/maintenance period and the aim is to recruit 25 patients.
The first patient began treatment in March 2023 with more due to begin soon.